
1. EXTRACTION OF GENOMIC DNA
For DNA extraction it is recommend using the reagents included in the DNA EXTRACTION KIT (MAD-
003951M). Before starting the DNA extraction, thaw out the reagents supplied in the kit: mineral oil, lysing
solution and a vial of protease solution. After use, the mineral oil and the lysing solution can be stored at 4ºC.
The protease solution must be stored at -20ºC, avoiding repeated freezing/thawing
1.1. SECTIONS OF PARAFFIN-EMBEDDED TISSUES
1. Take 1-4 tissue sections of 10 m thickness (according to the amount of material in each
section) and place in a 1.5 ml microcentrifuge tube using a needle or fine tweezers.
2. Add 500 μl of mineral oil (included in the kit) heat in a heating block for 2 min at 95ºC.
Centrifuge 2 min at 8000 rpm in the microcentrifuge.
3. Remove the oil without disturbing the tissue fragments.
4. Repeat steps 2 and 3. (The remaining mineral oil does not interfere with the DNA extraction
process).
5. Prepare a 50:1 mixture of lysing and protease solutions (for every 50 μl of lysing solution add
1 μl of protease solution) in sufficient volume for processing the tissue samples.
6. To the resulting tissue pellet add an adequate volume (50-500 μl) of the mixture above until
the tissue is fully in suspension (this is important to ensure a good yield of DNA and degrading of
contaminating cell remains which could interfere with subsequent amplification of this DNA).
7. Agitate several times using the micropipette to homogenize, centrifuge for 5 sec to remove
bubbles and incubate for 24-48 h at 55ºC in thermostatic bath or heating block.
8. Heat at 95ºC in the thermal block for 8-10 min to inactivate the protease.
9. Centrifuge for 5 min at maximum speed. After centrifugation there should be 2 phases, the upper
containing the remains of the mineral oil and lower aqueous layer that contains the dissolved
DNA. There may be a small pellet of undigested tissue at the bottom of the tube. COLLECT THE
AQUEOUS PHASE containing the DNA, avoiding any tissue remains at the bottom of the tube.
10. Use 3 μl of this DNA solution for amplification. The sample can be stored in this state, being
stable at 4ºC for 1 week, or at -20/-80ºC for several months.
1.2. FROZEN TISSUE
1. Cut 1-2 sections of tissue in the cryostat or using a scalpel blade and place in a 1.5 ml
microcentrifuge tube using a needle or fine tweezers.
2. Continue with step 5 of the previous protocol for paraffin sections.
Note: To avoid evaporation of the lysing/protease solution during the incubation at 55ºC, a few drops
of the mineral oil included in the kit can be added.
1.3 PERIPHERAL BLOOD OR BONE MARROW SAMPLES
Note: Do not use heparinized whole blood. We recommend the use of EDTA or sodium citrate as
anticoagulants.
1. Isolate the white cell population using the Buffy Coat from 1.5 ml of blood/marrow with EDTA.
Fractionate by centrifuging at 1500-2000 x g for 10-15 min at room temperature. This procedure
separates an upper phase of plasma, a lower phase with the red cell population and a thin interphase
with white cells (the buffy coat). (For a typical clinical centrifuge, 1500-2000 x g is equivalent to 3000-
3400 rpm).
2. Collect the white cell population and place it in a clean tube.
3. Wash with 5 ml of TE 1X buffer and incubate for 10 min at 37ºC to lyse any remaining
haemocytes.
4. Centrifuge at 3000 rpm for 5 min to precipitate white cells.
5. Re-suspend the cell pellet in 1 ml of TE 1X buffer and transfer to a 1.5 ml Eppendorf tube.
6. Centrifuge at 8000 rpm for 5 min in a microcentrifuge and discard the supernatant.
7. Continue with step 5 of protocol 1.1 for paraffin-embedded samples.
Note: To avoid evaporation of the lysing/protease solution during the incubation at 55ºC, a few drops of the
mineral oil included in the kit can be added.
2. AMPLIFICATION REACTION
For each analysis sample of test DNA, four mixtures are amplified: two for IgK, one for IgL and an
internal control mix (CI). The mixes should be protected from light since they contain fluorescence-labelled
primers.
2.1. SAMPLE PREPARATION
1. Thaw 1 blue tube (IgK-A), 1 orange tube (IgK-B), one green tube (IgL) and one yellow tube
(internal control) for each sample, and, keeping them on ice, add the following to each tube:
– 1 μl of enzyme Phire® Hot Start II DNA Polymerase
– 3 μl of the DNA sample*
2. Mix well and centrifuge for 5 sec in the microcentrifuge to remove bubbles.
*If the DNA is of known concentration, 200-500 ng of DNA per tube is recommended.
Note: It is important to keep the samples on ice until they are placed in the thermocycler to avoid nonspecific
binding of the primers.
2.2. AMPLIFICATION
1. Place all tubes in the thermocycler and amplify using the following programme:
98ºC 2 min.
35 cycles:
98ºC 10 s
60ºC 10 s
72ºC 15 s
72ºC 1 min
2. When the reaction is over, keep the tubes at 12-15ºC. If the samples are not to be processed
immediately, they can be stored at 4ºC or -20ºC until use.
2.3. RECOMMENDED AMPLIFICATION CONTROLS
The kit supplies clonal and polyclonal positive control DNAs, which should be amplified with each batch
of samples analyzed.
To control possible cross-contamination between samples or contamination of the common reagents used
during sample handling, we recommend the processing of a blank sample without DNA. This contains all
the same reagents employed in the DNA extraction from samples and is prepared using the same
processing procedures. Because the blank sample contains no DNA, it should give a negative result for all
three mixtures after amplification.
Likewise, although not essential, it is advisable to analyze each sample in duplicate, i.e., dividing the same
genomic DNA to carry out the amplifications of all fragments in duplicate. This facilitates interpretation of
doubtful results and confirms, using the duplicate, the size of monoclonal peaks in the profile of a reactive
polyclonal population.
3. ANALYSIS OF AMPLIFIED PRODUCTS
3.1. GEL ELECTROPHORESIS
WARNING! Given the high sensitivity of the amplification technique, which generates large quantities of a
specific DNA fragment, this amplified product represents a potent source of contamination in the laboratory.
It is recommended that the handling and electrophoresis of amplified products be carried out in a work area
well removed from the sample preparation area to avoid contamination by amplified DNA and consequent
false positive results.
Amplified products can be visualised by using 4% agarose or 6-8% polyacrylamide gels with half-strength
TBE buffer. Polyacrylamide gels give the best resolution.
Master Diagnóstica supplies “ready-to-use” kits for DNA electrophoresis in either agarose or polyacrylamide
gels, including all reagents needed for electrophoresis: molecular weight marker, loading buffer, 0.5 x TBE
buffer concentrate and EtBr (Cat. Nº MAD-003980M and MAD-003990M for agarose and polyacrylamide,
respectively).
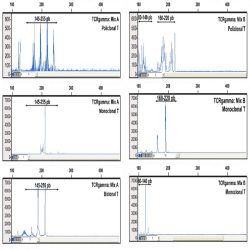
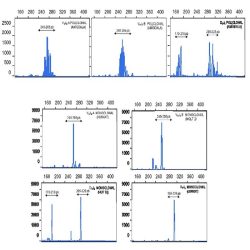
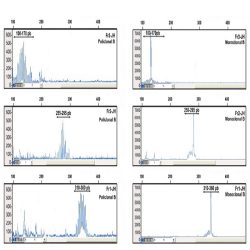
Heteroduplex analysis of the PCR products is recommended to differentiate between products derived from
mono and polyclonal populations. If there has been a clonal rearrangement of the IgK/IgL genes, a
“homoduplex” will be obtained, whereas in a polyclonal population, a “heteroduplex” with heterogeneous
binding will be formed after the denaturing/renaturing procedure.
3.1.1. PROCEDURE
1. Denature the PCR products at 94ºC for 5 min.
2. Renature by transferring rapidly onto ice (4ºC) for 10-60 min.
3. Assemble the agarose or polyacrylamide gel in its tray and cover with electrophoresis buffer TBE
0.5X.
4. Take 20 μl of PCR product and mix with 4 μl of loading buffer 6X.
5. Load the samples into the wells in the gel, placing 10 μl of molecular weight marker in one of the
tracks.
6. Carry out electrophoresis for 1-2 h at 100 V. Voltage and running time can vary depending on the type
of gel, the electrophoresis tray, and the size of the amplified product, among other factors.
7. Stain with 0.5 μg/ml EtBr in water or TBE 0.5X and visualize with a UV transilluminator.
3.2. CAPILLARY ELECTROPHORESIS
PCR mixes IgK-A, IgK-B, IgL and internal control mix, contain primers labelled with 6-FAM fluorochrome. In
addition to visualization by conventional electrophoresis, this allows the PCR products to be analyzed using
capillary electrophoresis.
Automatic sequencers based on capillary electrophoresis, e.g., ABI PRISM® 3100 and 3500 Genetic
Analyzers from LifeTechnologies, are systems with high levels of reproducibility and sensitivity in the
sequencing and analysis of genomic fragments. Their performance is superior to that of the majority of
sequence analyses based on the use of polyacrylamide gels. Moreover, these systems allow rapid and
accurate analysis of the results using specific software.
3.2.1. PREPARATION OF THE PCR PRODUCT
1. Mix in a 0.2-ml Eppendorf tube:
15 μl of deionised formamide + 0.5 l of 600 LIZ marker + 1 or 2 μl of amplified DNA (this volume of
amplified product can be changed if the fluorescent signal is outside the optimum range).
2. Homogenize with a pulse of the vortex mixer.
3. Denature at 95ºC for 5 min, chill to 4ºC and load into the sequencer.
Electrophoresis conditions:
Gel: POP7 polymer
Buffer: ANODE and CATHODE BUFFER CONTAINER 3500 SERIES
Electrophoresis: approx. 40 min and 2-6 injection time.
Optimum fluorescence intensity: up to a maximum of 10,000 units
3.2.2. OPTIMUM CONDITIONS FOR SAMPLE LOADING
To test whether the DNA sample has been processed correctly and is of adequate quantity and quality to
generate a valid result in the electropherogram, we recommended loading an aliquot of the PCR products
(including the internal control) and carrying out conventional electrophoresis with EtBr staining before the
capillary electrophoresis analysis.
An excessive load of PCR product in capillary electrophoresis can lead to artefact peaks for the molecular
weight standard and the product, leading to a false reading. Thus, excess of a clonal sample can produce
peaks that resemble the pattern for a polyclonal sample.
To avoid this problem, the fluorescent signal should be kept between 400 and 4000 fluorescence units in
models 3100 and 3500.
Reviews
There are no reviews yet.